


Sequencing Wasn't the Bottleneck
Last year, we were working with a mid-sized clinical genetics lab on a bioinformatics consulting project. During one of our discussions, their lead geneticist mentioned something that stuck with us: "We have a three-week backlog on variant interpretation." This wasn't a resource problem they could solve by hiring more people. Their small bioinformatics team was spending entire days running variants through different annotation tools, cross-referencing gnomAD frequencies, then manually looking up each significant variant in OMIM and PubMed. The whole process was taking two to four weeks per sample, and the backlog was growing.
At disease-targeted testing laboratories, even with semi-automated tools, variant assessment averages 40 minutes per variant, 22 minutes when literature review is excluded. Their analysts were dealing with hundreds of variants per exome, and the math gets painful fast. While sequencing itself has approached the "$1,000 genome" threshold, interpretation costs haven't followed the same curve. The infrastructure costs for making sense of that data remain substantial, and unlike sequencing, they're not falling.
The bottleneck in clinical genomics isn't generating data anymore. It's understanding what the data means.
We Built, What Clinicians Actually Need
We set out to answer a straightforward question: what if interpreting genomic data required uploading a file and clicking "Analyze"? The platform we developed handles the complete tertiary analysis workflow:
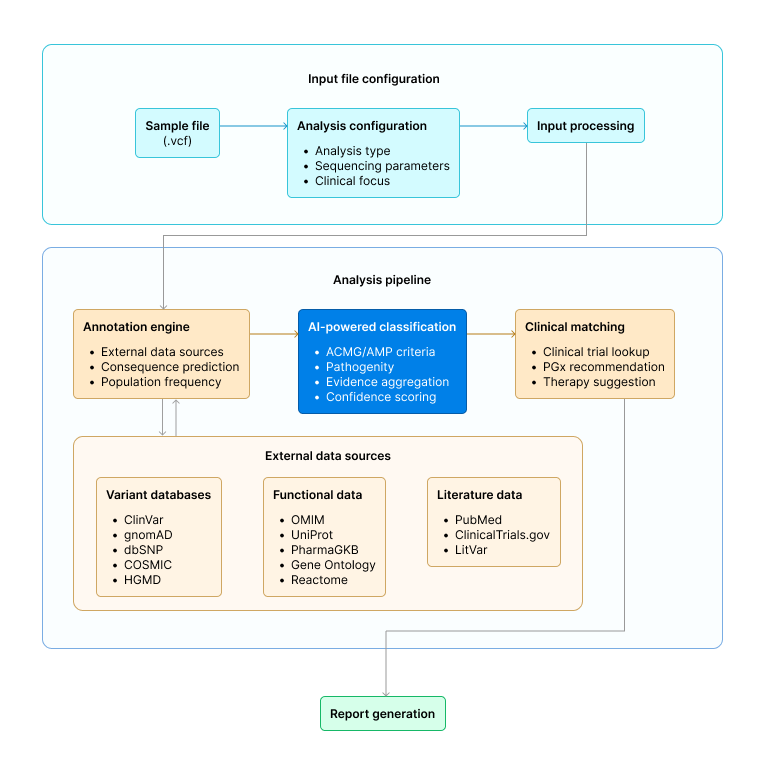
- Data Input. Clinicians upload a VCF file directly, no reformatting required.
- Analysis Selection. Pre-configured workflows cover whole exome sequencing (germline or somatic), whole genome sequencing, targeted gene panels (hereditary cancer, cardiac, neurological, or custom), and pharmacogenomics.
- Automated Annotation and Classification. The platform annotates variants across 10+ authoritative databases (ClinVar, gnomAD, dbSNP, COSMIC, PharmGKB, OMIM, HGMD), applies ACMG/AMP classification guidelines with evidence breakdown, scores pathogenicity using 12+ in silico predictors (SIFT, PolyPhen-2, CADD, REVEL, and others), identifies clinically actionable findings, matches variants to FDA-approved therapies and active clinical trials, and generates pharmacogenomic recommendations.
- Report Generation. A comprehensive clinical report in PDF, DOCX, or interactive HTML format, ready for patient consultation. Processing completes in 30–60 minutes.
The genetic counselor receives a clinical report in PDF, reviews it, and has it ready for the patient consultation within a few minutes instead of days. The entire process, from VCF upload to a report ready for a clinical conversation, takes less than an hour. If that same case, routed through the manual workflow, it would have taken two to three weeks.
Why This Matters Beyond Efficiency
Genomic medicine should be accessible to any clinic that wants to use it, not just major academic medical centers with specialized bioinformatics teams.
A small community hospital should be able to offer genomic testing to their cancer patients. A solo genetic counseling practice should be able to provide carrier screening without outsourcing to expensive lab services with month-long turnaround times. The technology for sequencing is already there, affordable and widespread. The bottleneck is interpretation. Remove that bottleneck, and genomic medicine becomes practical for the many patients who could benefit from it but don't currently have access to it.
This isn't about replacing clinical expertise. A platform doesn't make diagnostic decisions, clinicians do. What it does is collapse the weeks of annotation, database cross-referencing, and evidence gathering into a structured starting point, so the clinician's time goes toward the judgment calls that actually require their training.
Where We Can Take This Further
- AI-powered phenotype matching for rare disease diagnosis. Rare disease patients often wait years for diagnosis. Phenotype-driven variant prioritization can significantly reduce the time to identify causal variants.
- Trio analysis for de novo variant detection. Comparing proband, mother, and father genomes enables detection of de novo mutations that would be impossible to identify from singleton analysis.
- Structural variant interpretation. Inversions, translocations, and large copy number variants require different analytical approaches than SNVs and small indels.
- Polygenic risk scoring. For common diseases where risk is distributed across many variants of small effect, aggregate scoring provides clinically relevant risk stratification.
- EMR integration. Seamless clinical workflows require genomic results to flow directly into electronic medical records without manual transcription.
- Real-time collaboration tools. Multidisciplinary tumor boards and rare disease case conferences benefit from shared access to variant evidence and classification rationale.
References
- Schwarze K, Buchanan J, et al.. The complete costs of genome sequencing: a microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet Med. 2020 Jan;22(1):85-94. doi: 10.1038/s41436-019-0618-7. Epub 2019 Jul 30. PMID: 31358947; PMCID: PMC6944636.
- Attard CA, Carmany EP, Trepanier AM. Genetic counselor workflow study: The times are they a-changin'? J Genet Couns. 2019 Feb;28(1):130-140. doi: 10.1002/jgc4.1041. Epub 2018 Dec 10. PMID: 30629774.
- Castellanos E, Romero D, Martin A, et al. Comprehensive evaluation of ACMG/AMP-based variant classification tools. Bioinformatics. 2026;42(2):btaf623.
- Nicora G, Limongelli I, Gambardella G, et al. A machine learning approach based on ACMG/AMP guidelines for genomic variant classification and prioritization. Scientific Reports. 2022;12(1):2517.