
Run production-grade Nextflow pipelines using AWS Batch with minimal setup. No complexity. No orchestration headaches. Just working on pipelines.
What is Nextflow?
Before we dive into the “why AWS Batch” question, let’s talk about what Nextflow actually is.
Nextflow is a workflow orchestration engine designed for data-intensive computational pipelines. Think
of it as a sophisticated task manager that can:
-
Chain together multiple steps: Take raw data, run it through analysis tools, and
generate reports in a defined sequence
-
Handle dependencies automatically: Task B won’t run until Task A finishes
successfully
-
Parallelize work: If you have 100 samples to analyze, Nextflow can process them all
at once (resource permitting)
-
Resume from failures: If your pipeline crashes at step 5 out of 10, you can restart
from step 5 instead of starting over
Originally built for genomics (where a single
analysis might have 20+ steps and run for days), Nextflow is now popular in data science, ML pipelines,
and any field dealing with complex data processing.
The key feature? Nextflow utilises
containers (such as Docker) for each task, ensuring your analysis is reproducible; it’ll run the same
way on a laptop as it does on a cloud cluster.
Why AWS Batch for Nextflow?
AWS Batch
is a managed service that runs batch computing workloads. You define what to compute, AWS handles
everything else: provisioning servers, queuing jobs, scaling up and down, and cleanup.
Here’s
why it pairs perfectly with Nextflow:
-
Hands-off infrastructure: No servers to patch, no clusters to maintain
-
Smart scaling: Starts instances when jobs arrive, stops them when idle
- Flexible pricing: Choose on-demand for or spot for 90% cost savings
- Built-in monitoring: CloudWatch tracks everything automatically
- Simple architecture: Fewer moving parts means fewer things to break
Compare this to running your own compute cluster or managing complex infrastructure. AWS Batch
removes all that operational burden.
How it Works:
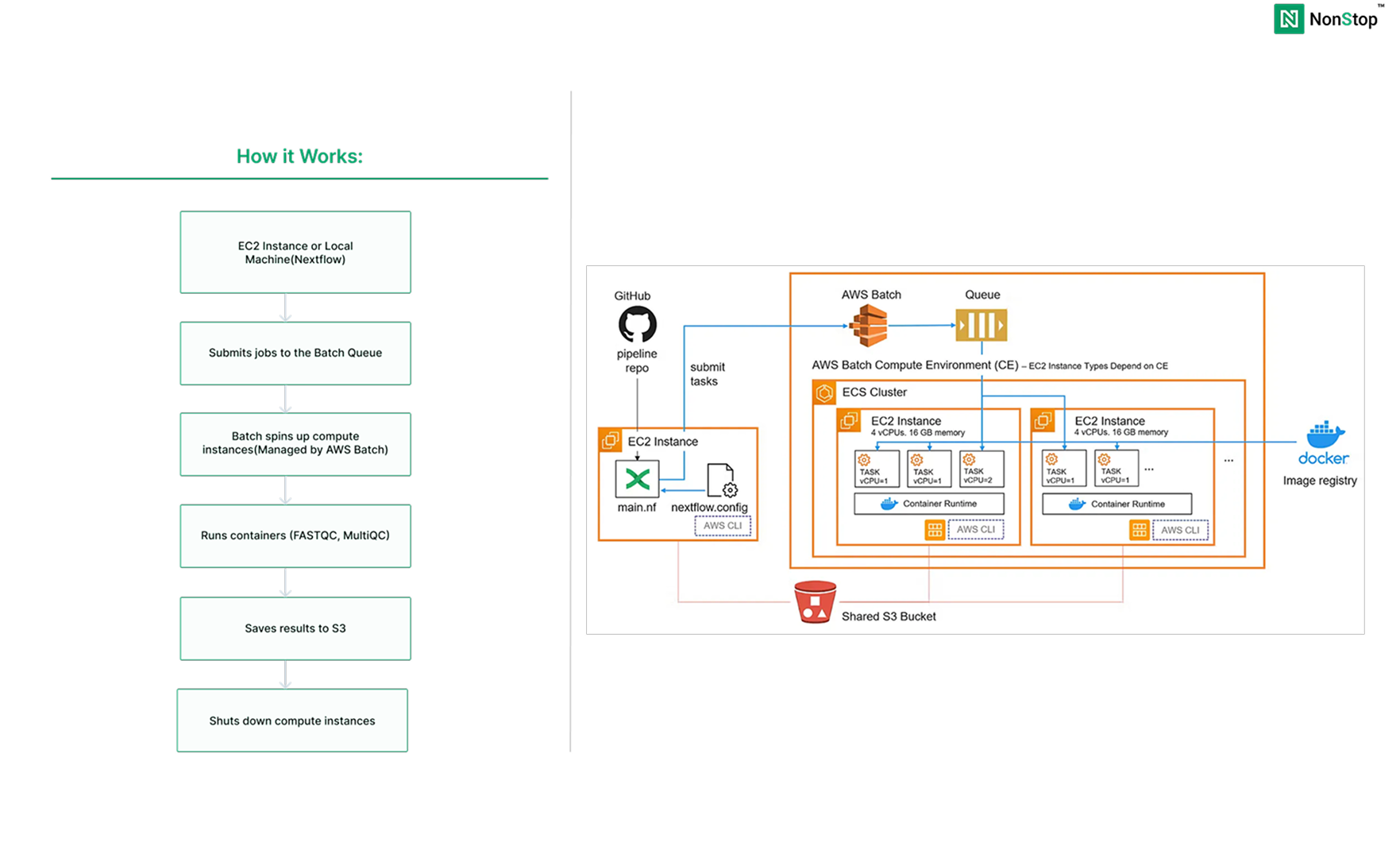
What We’re Building
- S3 bucket (stores input data, results, and temp files)
- Batch compute environment (the worker pool)
- Batch job queue (routes work to available compute)
- One EC2 instance (runs the Nextflow coordinator)
- The rest will be managed by AWS Batch
Building Our Infrastructure
Step 1: Create S3 Bucket
1. Open S3 Console:
- AWS Console → S3 → Create bucket
2. Configure:
1. Open S3 Console:
-
Bucket name:
nextflow-demo-yourname(must be globally unique) - Region: select your nearest region
- Keep defaults
- Click Create bucket
3. Create Folders:
Inside the bucket:
-
Click Create folder → Name:
inputs→ Create -
Create folder:
results -
Create folder:
work - Click Create bucket
4. Upload Data:
-
Open
inputs/folder - Click Upload
- Add your FASTQ file
- Click Upload
Step 2: Create IAM Roles
We need two roles: one for the EC2 instance, one for the Batch compute environment.
Role 1: EC2 Instance Role
1.Open IAM Console
- AWS Console → IAM → Roles → Create role
2. Select Trusted Entity:
- AWS service → EC2
- Click Next
3. Attach Policies
Add these three policies:
-
AmazonS3FullAccess -
AWSBatchFullAccess -
AmazonECS_FullAccess -
AmazonECS_FullAccess
Note: To restrict this to the exact bucket, you can create a custom policy and attach it
instead of the AmazonS3FullAccess policy.
Click Next
4. Name and Create:
- Role name:
NextflowEC2Role - Click Create role
Step 3: Create AWS Batch Compute Environment
1. Open Batch Console:
- AWS Console → Batch → Compute environments → Create
- Select Amazon EC2
2. Orchestration Type:
- Select Managed
3. Name:
-
Compute environment name:
nextflow-compute
4. Service Role:
- Select Create a new service role
- AWS creates this automatically
5. Instance Configuration:
-
Instance role: Select
BatchComputeInstanceRole - Click Next
6. Capacity
PS: You can modify this as per your requirements
-
Provisioning model: On-Demand or select Spot
- Minimum vCPUs: 0
- Desired vCPUs: 0
- Maximum vCPUs: 16
- Instance types: select default_x86_64
-
Allocation Strategy: keep it default, or you can select as per your
need
- Click Next
7. Network:
- Use the default VPC
- Select all available subnets
- Security groups: Default
8. Create
- Click Create compute environment
- Wait for status: VALID
Step 4: Create Job Queue
1. Navigate
- Batch → Job queues → Create
2. Configure
- Select Orchestration type: EC2
-
Job queue name:
nextflow-queue - Priority: 1
-
Connected compute environments: Select
nextflow-compute
3. Create
- Click Create job queue
Step 5: Launch EC2 Instance
1. Open EC2 Console:
- AWS Console → EC2 → Launch instance
2. Basic Configuration:
-
Name:
nextflow-launcher - AMI: Amazon Linux
- Instance type: t3.small
- Key pair: Select existing or create new
3. Network Settings:
- VPC: Default
- Auto-assign public IP: Enable
- Security group: Create new or use existing
- Allow SSH (port 22) from your IP
4. Advanced Details:
- Scroll down to IAM instance profile
-
Select:
NextflowEC2Role
5. Launch:
- Click Launch instance
-
Select:
NextflowEC2Role
Step 6: Install Nextflow
1. Connect to the Instance
If required, the pipeline can also be run from a local system after installing and configuring the AWS CLI. However, we’ve set up a dedicated EC2 instance for this purpose, and we’ll be using that going forward.
2. Connect to the Instance
1. Connect to the Instance
-
If required, the pipeline can also be run from a local system after installing and
configuring the AWS CLI. However, we’ve set up a dedicated EC2 instance for this
purpose, and we’ll be using that going forward.
2. Install Nextflow
sudo dnf install java -y
curl -s https://get.nextflow.io | bash
sudo mv nextflow /usr/local/bin/
sudo chmod +x /usr/local/bin/nextflow
nextflow -versionThe Nextflow Pipeline
What we’re doing:
-
FASTQC: Analyzes our raw sequencing data (FASTQ file) and generates
quality control reports. It checks things like read quality scores, sequence
duplication levels, GC content, and potential contamination. Each dataset gets its
own HTML report with pretty graphs.
-
MultiQC: Takes all those individual FASTQC reports and aggregates
them into a single, beautiful summary report. Instead of opening 10 different HTML
files, you get a comprehensive view of your data quality in one place.
Think of it this way: FASTQC is your detailed inspector, checking each piece individually, and MultiQC is the manager who summarises everything into a single executive report.
main.nf - This is your pipeline definition
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
params {
reads = 's3://your-bucket-name/inputs/sample_data.fastq'
outdir = 's3://your-bucket-name/results'
}
process FASTQC {
container 'biocontainers/fastqc:v0.11.9_cv8'
publishDir "${params.outdir}/fastqc", mode: 'copy'
input:
path reads
output:
tuple path("*.html"), path("*_fastqc.zip")
script:
"""
echo "Running FASTQC on ${reads}"
fastqc ${reads}
echo "FASTQC completed for ${reads}"
"""
}
process MULTIQC {
container 'multiqc/multiqc:dev'
publishDir "${params.outdir}/multiqc", mode: 'copy'
input:
path fastqc_reports
output:
path "multiqc_report.html"
path "multiqc_data"
script:
"""
echo "Aggregating FASTQC reports with MultiQC"
multiqc .
echo "MultiQC report generated successfully"
"""
}
workflow {
reads_ch = Channel.fromPath(params.reads)
fastqc_reports_ch = FASTQC(reads_ch)
MULTIQC(fastqc_reports_ch.collect())
}
nextflow.config - This tell Nextflow how to run on batch :
plugins {
id 'nf-wave'
id 'nf-amazon'
}
wave {
enabled = true
}
fusion {
enabled = true
}
process {
executor = 'awsbatch'
queue = '<job-queue-name>'
cpus = 1
memory = '2 GB'
// Error strategy: retry on transient AWS Batch issues
errorStrategy = { task.exitStatus in [130, 137, 143, 151] ? 'retry' : 'finish' }
maxRetries = 3
withName: FASTQC {
container = 'biocontainers/fastqc:v0.11.9_cv8'
}
withName: MULTIQC {
container = 'multiqc/multiqc:v1.14'
}
}
aws {
region = 'us-east-1'
batch {
volumes = '/fusion'
}
}
workDir = 's3://<your-bucket-name>/work'
Important: Replace
<yourbucket-name> and <job-queue-name>with your actual name in
both files.Push this to your GitHub repo.
Run the Pipeline
nextflow run https://github.com/username/repo-name.git
Outlook will look like:
N E X T F L O W ~ version 24.04.4
Pulling username/repo-name ...
downloaded from https://github.com/username/repo-name.git
Downloading plugin nf-wave@1.16.1
Downloading plugin nf-amazon@3.4.2
Downloading plugin nf-tower@1.17.3
Launching `https://github.com/username/repo-name` [awesome_lichterman] DSL2 - revision: e4b5f72abf [main]
executor > awsbatch (2)
[a1/b2c3] process > FASTQC [100%] 1 of 1 ✔
[e5/f6g7] process > MULTIQC [100%] 1 of 1 ✔
Completed at: 08-Jan-2025 10:30:15
Duration : 3m 45s
CPU hours : 0.4
Succeeded : 2
What happens:
- Nextflow submits jobs to AWS Batch
- Batch automatically spins up EC2 instances
- Runs FASTQC and MultiQC in containers
- Saves results to S3
- Shuts down instances when done
Conclusion
You’ve just built a production-ready Nextflow pipeline on AWS Batch. With minimal setup (S3, two IAM
roles, Batch environment, and one EC2 instance), you now have a system that:
- Automatically scales compute based on workload
- Handles job queuing and execution without manual intervention
- Saves results directly to S3
- Costs only pennies per run
The beauty of this setup is its simplicity. No complex networking. No multiple services to maintain.
Just Nextflow coordinating jobs and AWS Batch handling the compute.
What makes this powerful:
AWS Batch removes the operational burden. You don’t patch servers, manage clusters, or worry about
capacity planning. Submit your pipeline, and AWS takes care of the rest.
Nextflow handles the complexity of your workflow. Dependencies, retries, parallelization, and resume
capability are all built in. Your job is defining what to analyze, not how to manage the
infrastructure.
Real-world impact:
This same setup scales from analyzing a single sample to processing thousands. The pipeline we built
today (FASTQC + MultiQC) can easily extend to variant calling, RNA-seq analysis, or any multi-step
computational workflow.
Teams using this approach report:
- 90% reduction in infrastructure management time
- 80–90% cost reduction on spot instance
- Faster iterations on analysis pipelines
- Reproducible results across different environments
Happy Hosting!